21 CFR 801 vs. 21 CFR 803: Understanding Labelling and Reporting Regulations



Summary
- 21 Code of Federal Regulations (CFR) 801 ensures proper labelling of medical devices.
- 21 CFR 803 monitors the safety of medical devices.
- Manufacturers must comply with labelling requirements.
- Importers, user facilities, and manufacturers must report adverse events (AEs) or device malfunctions that could cause or contribute to serious injuries or death.
Short Description
The 21 CFR 801 and 21 CFR 803 provide medical device regulations and cover labelling and Medical Device Reporting (MDR) requirements, respectively.
What Is 21 CFR 801?
21 CFR 801 pertains to labelling, which provides safety and performance-related information to users and helps identify individual medical devices.
Manufacturers must specify this information on the label when the device is on the market, either on the device, on the packaging (or as a packaging insert), or as instructions for use.
Subparts Under the 21 CFR 801 Regulation
The various subparts under the 21 CFR 801 regulation include subparts A to H, which are described below:
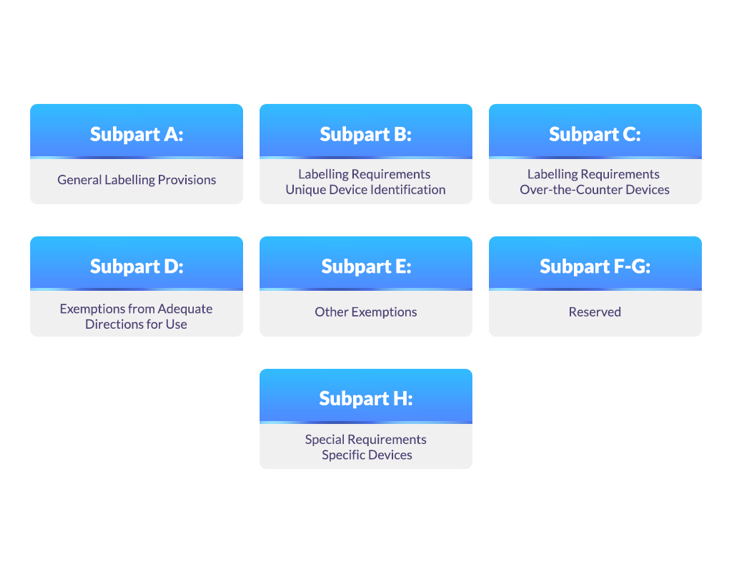
Figure 1: Subparts Under the 21 CFR 801
1. Subpart A-General Labelling Provisions:
This section discusses what a medical device label must include, such as the manufacturer’s, packer’s, and distributor’s name and place of business and adequate directions for use.
It also addresses the meaning of intended uses, any misleading statements, the use of symbols in labelling, the prominence of required label statements, and the format of dates to be used on the label.
2. Subpart B-Labelling Requirements-Unique Device Identification:
Medical device labels must include a unique device identifier (UDI); however, there are exceptions in certain cases. If a device qualifies for such exceptions, voluntary labelling can be done.
This section also mentions the technical requirements of every UDI, the labelling requirements of stand-alone software and requests for exceptions. Permanent marking is mandatory for the devices to be reused or reprocessed.
3. Subpart C-Labelling Requirements-Over-the-Counter Devices:
The area of the principal display panel (part of the label likely to be displayed) is mentioned to obtain a uniform type size for all same-size packages.
The identity of the commodity and the net quantity of contents must be declared. Devices made with or containing chlorofluorocarbons or other class I ozone-depleting substances must include warning statements.
4. Subpart D-Exemptions from Adequate Directions for Use:
Certain medical devices, like prescription devices, in vitro diagnostic products and those with commonly known directions are exempt from labelling adequate directions for use.
5. Subpart E-Other Exemptions:
This section elaborates on labelling and packaging exemptions that apply under specific circumstances when medical devices are processed, labelled, or repacked in substantial quantities at an establishment in a location other than the previous location packaging or processing.
6. Subpart F-G:
These sections are reserved.
7. Subpart H-Special Requirements-Specific Devices:
This section discusses labelling requirements for specific devices including devices equipped with impact-resistant lenses; devices that emit ozone; devices with chlorofluorocarbon propellants; articles for the lay public in the repairing/refitting of dentures; prescription hearing aids, menstrual tampons, latex condoms and devices containing natural rubber; and warning statements for prescription and restricted devices.
What Is 21 CFR 803?
MDR is a post-marketing surveillance tool that allows the Food and Drug Administration (FDA) to monitor device performance, detect safety issues, assess benefits and risks, avoid misbranded or adulterated devices and ensure devices remain safe and effective for their intended use.
CFR 803 outlines the requirements for importers, manufacturers, distributors and user facilities to report AEs and problems associated with devices to the FDA.
Subparts Under the 21 CFR 803 Regulation
The various subparts under the 21 CFR 803 regulation are mentioned in Figure 1.
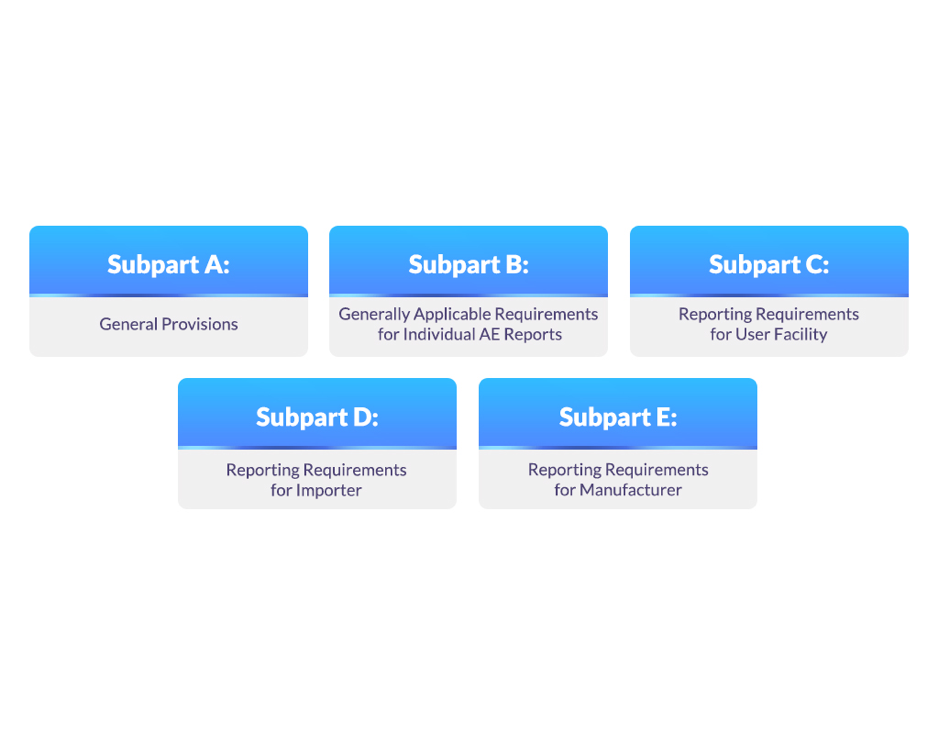
Figure 2: Subparts Under the 21 CFR 803
How to Report?
The FDA receives over 2 million per year MDRs of suspected device-associated serious injuries and deaths, as well as device malfunctions. The procedures for reporting are different for importers, manufacturers and user facilities as given below:
1. Importers:
Importers are required to report to both the manufacturer and the FDA within 30 calendar days if they become aware that one of their devices may have caused or contributed to a serious injury or death.
If a device malfunctions and poses a risk of leading to such outcomes, the importers must report it solely to the manufacturer.
2. Manufacturers:
Manufacturers must report to the FDA within 30 calendar days if they become aware that their devices may have caused or contributed to a serious injury or death. They must also report any malfunctions that could lead to such outcomes if they were to occur again.
3. User Facilities:
A hospital, nursing home, ambulatory surgical facility, or outpatient diagnostic/treatment facility, except a physician’s office, is a user facility. These facilities must report any suspected medical device-related death to both the manufacturer and FDA within 10 working days.
Serious injuries caused by a medical device must be reported to the manufacturer or FDA (if the manufacturer is unknown) in 10 working days. Reporting device malfunctions is not mandatory, but it can be done voluntarily.
21 CFR 801 vs. 21 CFR 803: Key Differences
While 21 CFR Part 801 and 21 CFR Part 803 serve different regulatory functions, they are interconnected in ensuring medical device safety and compliance. Table 1 enlists a detailed comparison of these two.
Table 1: 21 CFR 801 vs. 21 CFR 803
| 21 CFR 801 (Labelling Requirements) | 21 CFR 803 (Medical Device Reporting) | |
| Purpose | Guides manufacturers regarding the labelling of medical devices. | Monitors safety by reporting AEs and device-related problems. |
| Primary audience | Manufacturers | Importers, manufacturers and user facilities. |
| Scope | Provides safety information and instructions for use. | Monitors the safety and effectiveness of the device. |
| Compliance Requirement | Labels must inform users regarding the purpose, directions for use and risks or warnings of the device. | AEs and device problems that contribute to or cause a serious injury or death must be reported to the FDA within the stipulated time. |
| Preventive vs. Reactive | Preventive, as it ensures precise labelling to avoid potential harm/misuse before the devices are on the market. | Reactive, as it ensures safety by addressing AEs and device-related problems after devices are on the market. |
| Role in Public Safety | Reduces the risks of improper handling and misuse. | Prevents further injuries or death. |
*AE: Adverse event; CFR: Code of Federal Regulations; FDA: Food and Drug Administration
Conclusion
21 CFR 801 ensures proper labelling of medical devices to inform users regarding their safety and performance-related information, while 21 CFR 803 monitors safety by reporting any AEs and device-related issues.
Due to the high number of MDRs of suspected device-associated serious injuries and deaths, it is essential to understand and comply with these regulations to ensure medical devices are safe, effective and user-friendly.
References
- Labelling for Medical Devices. Global Harmonization Task Force. [cited 2025 February 06]. Available from: https://www.imdrf.org/sites/default/files/docs/ghtf/final/sg1/technical-docs/ghtf-sg1-n43-2005-labelling-medical-devices-050603.pdf
- Labeling. Electronic Code of Federal Regulations. [cited 2025 February 06]. Available from https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-801?toc=1
- Medical Device Reporting (MDR): How to Report Medical Device Problems. Food and Drug Administration. [cited 2025 February 06]. Available from https://www.fda.gov/medical-devices/medical-device-safety/medical-device-reporting-mdr-how-report-medical-device-problems
- Medical Device Reporting. Electronic Code of Federal Regulations. [cited 2025 February 06]. Available from https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-803#803.1
Neha Gupta
CliniExperts Services Pvt. Ltd.
Recent Posts
Global Regulatory Strategy for Medical Devices – A CliniExperts Guide

This Article is all about Global Regulatory Strategy for Medical Devices – A CliniExperts Guide Introduction Medical Device manufacturers who are planning to enter the global market to market ..
The Role of US FDA 510(k) Consultants in Medical Device and IVD Approvals

Summary Short Description Navigating the U.S. Food and Drug Administration’s (FDA) 510(k) premarket notification process is a critical step for medical device and in vitro diagnostic (IVD) manuf..
Contact us
Please feel free to talk to us if you have any questions. We endeavour to answer within 24 hours.