EU Regulatory Strategy for Medical Devices: Pathways, Requirements & Market Access


Summary:
- Medical devices should be classified into risk categories (Class I, IIa, IIb, III) under the European Union Medical Device Regulation (EU MDR).
- Non-EU manufacturers must appoint an EU Authorised Representative (EU AR) to act as their regulatory contact in the EU.
- Manufacturers must undergo conformity assessment, often conducted by a Notified Body, to verify regulatory compliance.
- Technical documentation and a quality management system must be prepared to demonstrate device safety and performance.
- Clinical evaluation is required to support the device’s safety and effectiveness.
- Manufacturers must establish post-market surveillance systems to monitor device performance after market entry.
- Conformité Européenne (CE) marking confirms compliance with EU regulatory requirements and allows devices to be marketed across EU Member States.

An effective European Union (EU) regulatory strategy for Medical Devices involves a series of structured steps to ensure compliance with Regulation (EU) 2017/745. Manufacturers must address key requirements such as device classification, regulatory representation, conformity assessment, technical documentation, clinical evaluation, post-market surveillance, and Conformité Européenne marking. These elements collectively ensure that Medical Devices meet EU safety and performance standards before being placed on the market and continue to be monitored throughout their lifecycle.
Ensuring Compliance in the EU Medical Device Market
The European Union (EU) has established one of the most robust regulatory frameworks for Medical Devices, designed to ensure high standards of safety, performance, and transparency. The current system introduces stricter requirements for clinical evidence, device traceability, and lifecycle monitoring compared with earlier directives.1
This article outlines the EU regulatory strategy for Medical Devices, regulatory requirements, and compliance considerations for Medical Device manufacturers based on official EU regulatory guidance.
Overview of the EU Medical Device Regulation
The EU regulatory framework for Medical Devices is primarily based on Regulation (EU) 2017/745, commonly referred to as the European Union Medical Device Regulation (EU MDR). This regulation replaced earlier directives and introduced stronger oversight of device safety, performance, and post-market monitoring. The MDR framework aims to ensure that Medical Devices placed on the EU market are safe, effective, and supported by robust clinical evidence, while also promoting innovation and transparency.2
Key Elements of an EU Regulatory Strategy
A successful regulatory strategy requires manufacturers to address several key elements throughout the Medical Device lifecycle to ensure EU Medical Device compliance.
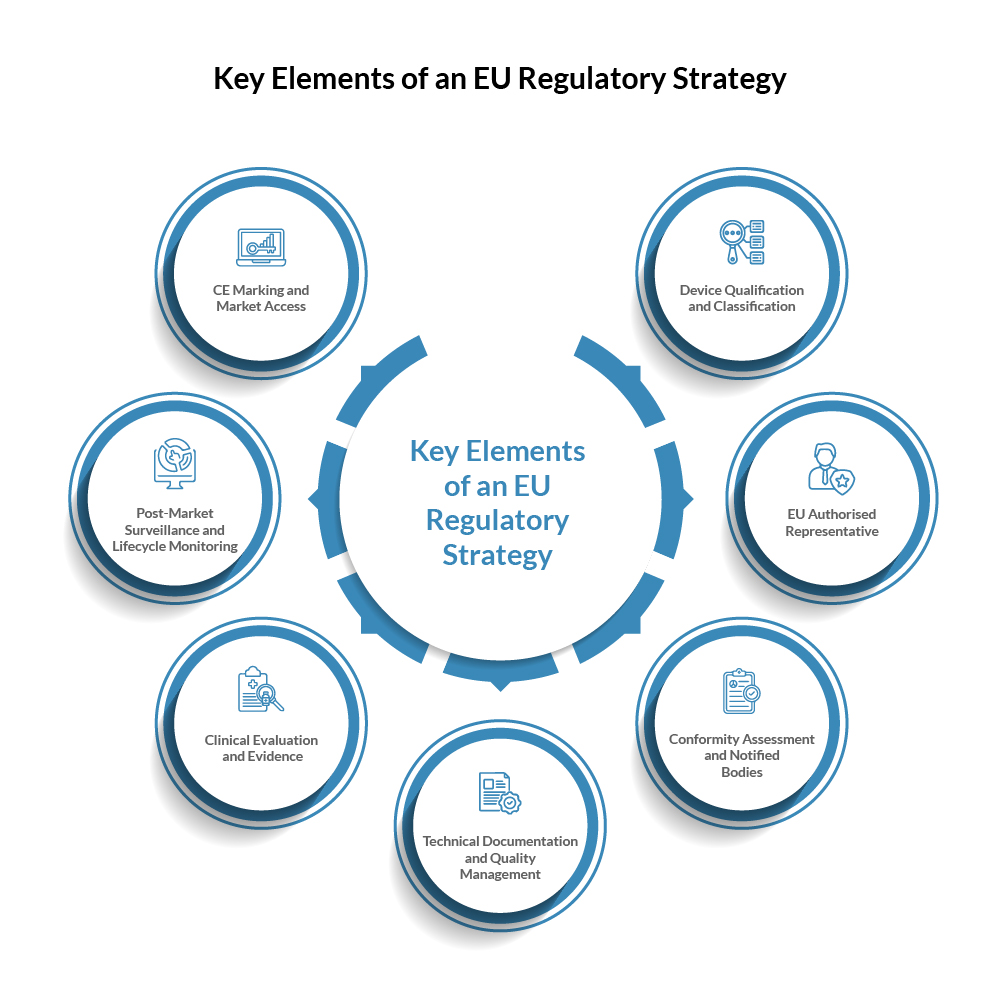
1.Device Qualification and Classification
The first step in a regulatory strategy is determining whether a product qualifies as a Medical Device and identifying its risk classification under Article 51 and Annex VIII of Regulation (EU) 2017/745.3 Under the MDR, devices are classified into four risk use based on risk during use (Table 1).4 The classification determines the level of regulatory scrutiny and conformity assessment procedures required before Medical Device market access in Europe.
| Device Class | Risk Level | Examples of Devices |
| Class I(Class Is, Im & Ir) | Low risk | Non-sterile surgical instruments, bandages, wheelchairs, stethoscopes |
| Class IIa | Low to Medium Risk | Dental fillings, infusion pumps, hearing aids, surgical gloves |
| Class IIb | Medium to high risk | Ventilators, orthopaedic implants, long-term contact lenses, infusion pumps for critical use |
| Class III | High risk | Pacemakers, heart valves, implantable defibrillators, breast implants |
Table 1: Risk classification of Medical Devices under the EU Medical Device regulation.
2. EU Authorised Representative
Manufacturers located outside the European Union must appoint an EU Authorised Representative (EU AR) to place Medical Devices on the EU market. The EU AR acts on behalf of the manufacturer in fulfilling certain regulatory obligations and serves as a liaison between a non-European Union manufacturer and the competent authorities.
Under Regulation (EU) 2017/745, the authorised representative is responsible for verifying that the technical documentation and declaration of conformity have been prepared, maintaining regulatory documentation, and cooperating with authorities in case of regulatory inquiries or safety issues.5
3.Conformity Assessment and Notified Bodies
An important step in the regulatory pathway is Notified Body selection, as manufacturers must choose an appropriate designated organisation to conduct the conformity assessment required under EU MDR.6
The Notified Body evaluates whether the device meets the regulatory requirements outlined in the MDR, including safety, performance, and quality standards.
4.Technical Documentation and Quality Management
Manufacturers must prepare extensive technical documentation demonstrating device safety and performance.3 This documentation typically includes:7
- Manufacturer details, including the name and address of the manufacturer or any authorised representative
- Product description providing a clear overview of the Medical Device
- Product identification, such as serial numbers or model numbers or UDI
- Design and manufacturing information, including the names and addresses of facilities involved in Manufacturing of the products.
- Notified Body information, if a Notified Body is involved in conformity assessment
- Conformity assessment procedure to be followed to demonstrate regulatory compliance
- Applicable EU regulatory requirements, including identification and analysis of relevant guidelines and essential requirements
- Technical standards used to demonstrate compliance (e.g., EN, ISO, and IEC)
- Risk management documentation, detailing identified risks and mitigation strategies
- Evidence of compliance, including testing reports for critical components or materials
- Product labelling and Instructions for Use (IFU)
In addition, manufacturers must maintain a quality management system to ensure continuous compliance throughout the device lifecycle.
5.Clinical Evaluation and Evidence
The EU MDR places strong emphasis on clinical evidence supporting the safety and effectiveness of Medical Devices. Manufacturers must conduct a clinical evaluation based on clinical investigations, scientific literature, or existing clinical data. For higher risk devices, additional clinical investigations may be required to demonstrate clinical performance and patient safety.8
6.Post-Market Surveillance and Lifecycle Monitoring
The MDR adopts a lifecycle approach to regulation, meaning that compliance does not end once a device enters the market. Manufacturers must implement:3,9
- Post-market surveillance systems
- Post-market clinical follow-up (PMCF)
- Incident reporting and vigilance procedures
These measures help regulators monitor device performance and address potential safety issues after market entry.
7.CE Marking and Market Access
Conformité Européenne (CE) marking indicates that a Medical Device meets the EU’s regulatory and safety requirements. Once CE marked, the device can generally be marketed across all EU Member States, enabling access to the EU’s single market for medical technologies.3 The CE marking process generally involves the following steps:10
- Identify applicable EU requirements for the product.
- Determine the conformity assessment procedure, including whether a Notified Body is required.
- Prepare technical documentation demonstrating compliance.
- Complete product evaluation and conformity assessment.
- Issue the conformity assessment EU MDR (EU Declaration of Conformity).
- Affix the CE marking, allowing the device to be marketed across the EU.
Conclusion
The EU regulatory requirements in healthcare for Medical Devices are built around the framework established by Regulation (EU) 2017/745. This regulation emphasises patient safety, stronger clinical evidence, and continuous lifecycle monitoring of Medical Devices.
For manufacturers aiming to enter the European market, understanding device classification, conformity assessment procedures, clinical evaluation requirements, and post-market surveillance obligations is essential for achieving regulatory compliance and securing CE marking. Thus, to effectively address these regulatory requirements, many companies seek EU MDR consulting services.
References
1. Chinmai B, D P. EU MDR Regulatory Update and Compliance Strategies. Asian J Pharm Res Dev. 2025;13(6):54-60. doi:10.22270/ajprd.v13i6.1652
2. New Regulations – Public Health – European Commission. February 3, 2026. Accessed March 9, 2026. https://health.ec.europa.eu/medical-devices-sector/new-regulations_en
3. French-Mowat E, Burnett J. How are Medical Devices regulated in the European Union? J R Soc Med. 2012;105(Suppl 1):S22-S28. doi:10.1258/jrsm.2012.120036
4. Guidance on classification of Medical Devices. MDCG 2021-24. Accessed March 9, 2026. https://health.ec.europa.eu/system/files/2021-10/mdcg_2021-24_en_0.pdf
5. Factsheet for Authorised Representatives, Importers and Distributors of Medical devices and in vitro diagnostic Medical devices. European Commission. Accessed March 9, 2026. https://health.ec.europa.eu/document/download/37879254-c7d6-487b-8fd9-06496e771465_en
6. Notified bodies for Medical Devices – Public Health – European Commission. February 3, 2026. Accessed March 9, 2026. https://health.ec.europa.eu/medical-devices-topics-interest/notified-bodies-medical-devices_en
7. Preparing technical documentation. Your Europe. Accessed March 9, 2026. https://europa.eu/youreurope/business/product-requirements/compliance/preparing-technical-documentation/index_en.htm
8. Oltmanns E, D’Agosto M, Spitzenberger F. “Appropriateness” of Clinical Data Under Regulation (EU) 2017/745– A Case Study and Survey. Ther Innov Regul Sci. 2025;59(6):1356-1368. doi:10.1007/s43441-025-00827-6
9. Pannonhalmi Á, Sipos B, Kurucz RI, Katona G, Kemény L, Csóka I. Advancing Regulatory Oversight of Medical Device Trials to Align with Clinical Drug Standards in the European Union. Pharmaceuticals. 2025;18(6). doi:10.3390/ph18060876
10. CE marking – obtaining the certificate, EU requirements. Your Europe. Accessed March 9, 2026. https://europa.eu/youreurope/business/product-requirements/labels-markings/ce-marking/index_en.htm
Recent Posts
Technical Documentation Requirements Under EU MDR and IVDR

Technical documentation is a fundamental requirement under the European Union Medical Device Regulation (EU MDR 2017/745) and In Vitro Diagnostic Medical Device Regulation (EU IVDR 2017/746). It provi..
Step-by-Step CE Marking Process for Medical Devices in the EU

CE Marking is a mandatory regulatory requirement for Medical Devices marketed in the European Union (EU) and European Economic Area (EEA). It confirms that a device meets necessary safety standards, h..
What is MHRA? Role, Responsibilities & Regulatory Framework Explained

MHRA is the UK’s official regulatory body that oversees medicines, vaccines, and Medical Devices to ensure they meet strict standards of safety, quality, and effectiveness. It evaluates products bef..
Contact us
Please feel free to talk to us if you have any questions. We endeavour to answer within 24 hours.