Gain a comprehensive understanding of the European Union Medical Device Regulation (EU MDR)


Summary:
- European Union Medical Device Regulation is a new set of regulations enforced in yearly 1990s.
- EU MDR primarily regulates medical devices in European countries.
- EU MDR mandates the manufacturers to demonstrate compliance and provide clinical data on the medical device’s safety and effectiveness to gain approvals.
- Medical devices may be any instrument, appliance, apparatus, software, reagent, material, implant, etc that can be used for humans for several medical purposes.
- Medical devices are classified into four different categories based on the risks associated with each medical device.
- Manufacturers must comply with the EU MDR requirement. This can be achieved by understanding the EU MDR in detail and by applying this understanding and building the MDR-compliant background.

European Union Medical Device Regulation
Introduction
European Union Medical Device Regulation or EU MDR is a set of regulations that regulate the production, distribution, sale and clinical investigation of medical devices in European countries. Before EU MDR, the Medical Devices Directives (MDD) were used to govern medical devices in Europe. Medical Devices manufacturers that intend to sell their medical devices in the European markets must mandatorily comply with EU MDR requirements.
The EU MDR mandates the manufacturers to demonstrate compliance and in-detail clinical information about device safety and performance efficiency which allows to gain approvals for each medical device.
European Union Medical Devices Regulation (EU MDR)
EU MDR was enforced to lay down certain rules relating to placing medical devices on the European markets for human use. EU MDR is also applicable to the clinical investigations related to such medical devices and accessories conducted in the European Union.
Reasons for replacing Medical Device Directives (MDD)
- MDD were indeed robust and innovative however, it had numerous loopholes in implementing medical devices safety, reliability and product quality.
- It was exposed by notorious incidents like the metal-on-metal hip scandal. Due to this event, the legislators were obliged to implement reform.
Medical Device Classification
Medical Devices may be defined as any instrument, appliance, apparatus, software, reagent, material, implant, or other article intended to be used by the manufacturer, alone or in combination, for human beings for one or more medical purposes.
Medical Devices may be used for diagnosis, prevention, monitoring, prognosis, treatment, in-vitro examination of specimens, etc.
Medical Devices may be classified into different categories.
- Class I medical devices are low-risk devices. These are non-invasive that do not interact with the body. Examples: hospital beds, bed pans, sterile plasters, etc.
- Class II a medical devices are medium-risk devices. These are generally invasive, however, limited to cavities/bodily openings. Examples: Thermometers, wheelchairs, hearing aids, weighing scales, ultrasonic diagnostic equipment, etc.
- Class II b medical devices are medium-risk devices. These are mostly used as surgical invasive/active devices partially or implantable in the human body. These may modify body fluids composition. Examples: infusion pumps, surgical lasers, ventilators, etc.
- Class III medical devices are high-risk medical devices. These are mainly used to support or sustain human life. These are important for preventing human impairment, illness or major injury. These may be used to connect to the brain or Central Circulatory System that contains medical products. Examples: Body implants such as vascular and neurological implants, heart valves, breast implants, cerebella stimulators, etc.
Scope of EU MDR
The new rules of MDR aim to reduce future incidents.
- These rules will enhance the requirement of proving the safety and efficacy of all medical devices.
- MDR will cover heavy medical devices used for liposuction, brain stimulators and lasers used for tattoo removal.
- Such products require certain specifications to market them which is now taken into consideration by the European Commission.
- MDR also covers medical device software, health application and machine-learning medical devices.
Challenges in implementing the MDR
Implementing the MDR in European countries may pose a number of challenges.
- MDR implementation may have increased the burden of compliance.
- It may have increased the time and cost of certification.
- The capacity of the notified body to assess is limited and may worsen in the run-up to May 2024.
- All the products which were marketed under the MDD may not proceed to market under these new regulations; this may challenge the device available in the market.
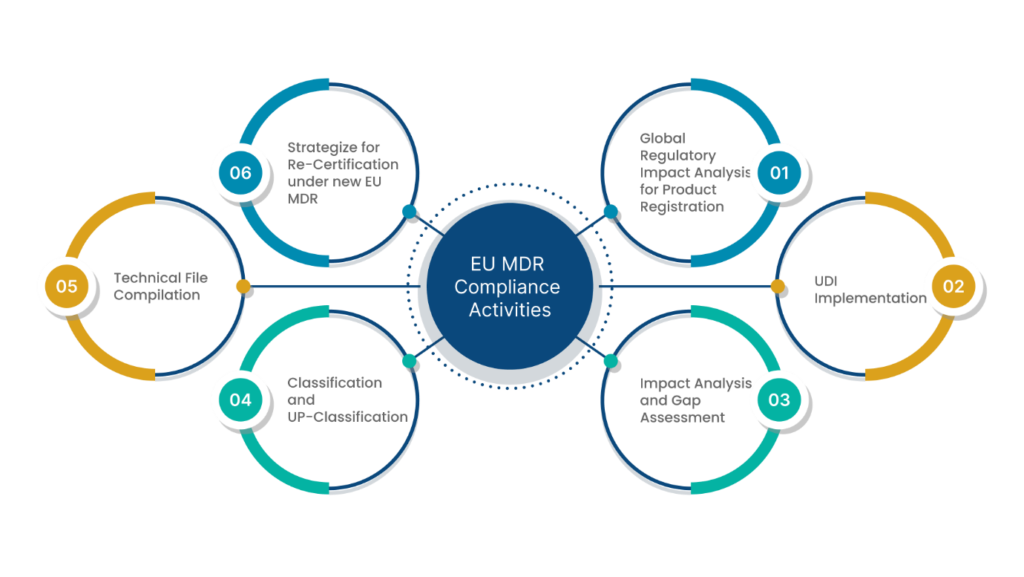
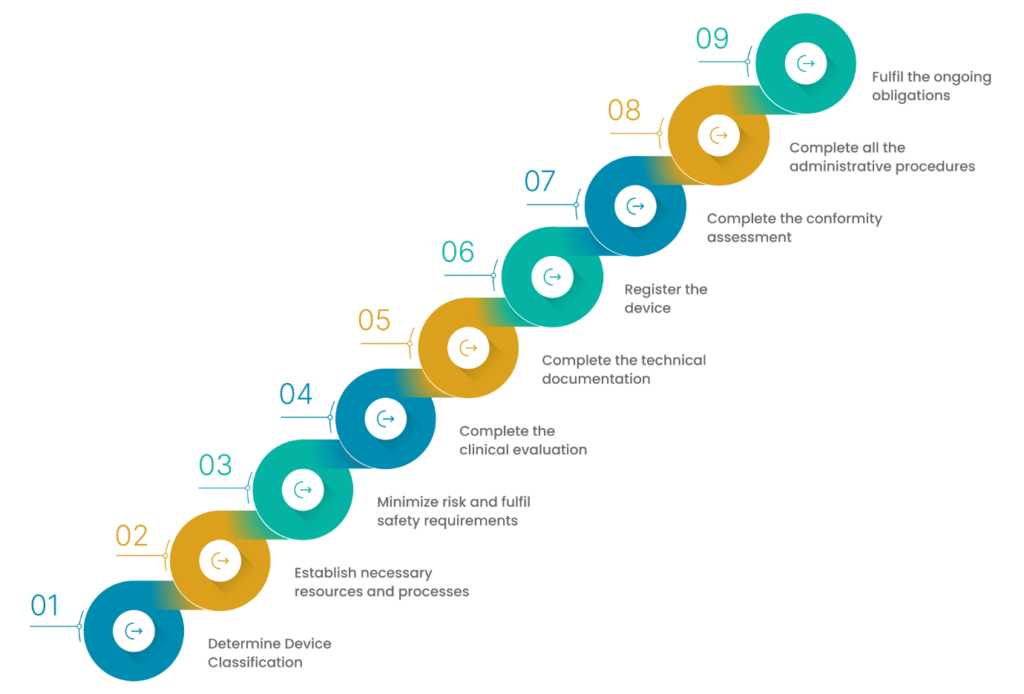
EU MDR Compliance
There are a few effective ways by which you can achieve EU MDR compliance. This can be done in two different phases:
1. Understanding the EU MDR in detail
2. Applying the understanding to build an MDR-compliant framework
EU MDR is a complex legislation that represents the largest part of medical device regulation. MDR has introduced a wide range of change that has placed a higher burden on manufacturers in several areas.
- MDR has broadened the definition of medical devices. It also covers medical software applications or nanomaterials which have been recently included for the first time in medical device legislation.
- MDR mandates the manufacturers to provide conventional clinical evidence through PMCF systems that demonstrate the safety and effective performance of a medical device.
- It has reduced the ability to claim equivalence. Under MDR the manufacturers cannot claim new devices as equivalent to already approved devices for sale. It requires its clinical evidence portfolio for sale.
- Change in the risk classification of some categories of medical devices has increased the regulatory burden on manufacturers.
- MDR emphasis on clinical evidence and post-market surveillance.
- Under MDR, there are only 38 Notified bodies which have gained approval for performing conformity assessments for medical devices.
Reference
- Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC[Internet]. [cited 2023 Jul 9]. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02017R0745-20170505
- Q&A: Medical Devices Regulation [Internet]. [cited 2023 Jul 3]. Available from: https://ec.europa.eu/commission/presscorner/detail/en/qanda_23_24
- Melvin T. LAW, ETHICS, AND POLICY The European Medical Device Regulation-What Biomedical Engineers Need to Know. [cited 2023 Jul 9]; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9395138/pdf/jtehm-melvin-3194415.pdf
- Achieve EU MDR and UKCA medical device compliance – Free Guide 2022 [Internet]. [cited 2023 Jul 3]. Available from: https://www.mantrasystems.co.uk/eu-mdr-compliance/
- French-Mowat E, Burnett J. How are medical devices regulated in the European Union? [cited 2023 Jul 9]; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3326593/pdf/JRSM-12-0036.pdf
CliniExperts - Your reliable partner for Comprehensive Compliance Solutions. We offer 360 degree Global Regulatory Solutions related to Pharma, Medical Devices and In-Vitro Diagnostics.
CliniExperts
CliniExperts Services Pvt. Ltd.
Recent Posts
What is MHRA? Role, Responsibilities & Regulatory Framework Explained

MHRA is the UK’s official regulatory body that oversees medicines, vaccines, and Medical Devices to ensure they meet strict standards of safety, quality, and effectiveness. It evaluates products bef..
EU MDR IFU Requirements: What Must Be Included for Compliance

IFU requirements (EU MDR) define what a Medical Device instructions leaflet must contain to ensure safe and correct usage of the Device. An IFU must contain all essential safety details, technical gui..
GDUFA Self-Identification: Complete FDA Compliance Guide (2026)

GDUFA self-identification is a mandatory FDA compliance process for generic Drug facilities and related sites. It supports FDA self-identification (Generic Drugs) by mandating structured annual submis..
Contact us
Please feel free to talk to us if you have any questions. We endeavour to answer within 24 hours.