GDUFA Self-Identification: Complete FDA Compliance Guide (2026)


Summary:
- Generic Drug user fee amendments (GDUFA) self-identification requires all eligible Generic Drug facilities and related sites to complete annual reporting with the U.S. Food and Drug Administration (FDA).
- The requirement applies to manufacturing, packaging, testing, and clinical research sites involved in generic Drug submissions under FDA regulations.
- Facilities must submit structured and standardized data using the Structured Product Labelling (SPL) format for accurate regulatory tracking.
- Only facilities involved in manufacturing Active Pharmaceutical Ingredients (APIs) and Finished Dosage Forms (FDFs) are required to pay applicable GDUFA facility fees.
- Failure to comply with GDUFA self-identification requirements can result in regulatory actions, including misbranding, legal consequences, and restrictions on product distribution.

GDUFA self-identification is a mandatory FDA compliance process for generic Drug facilities and related sites. It supports FDA self-identification (Generic Drugs) by mandating structured annual submissions. The process ensures transparency, supports regulatory oversight, and improves Drug approval timelines. Following proper submission steps and fee requirements helps avoid penalties, maintain legal product status, and ensure uninterrupted access to the US generic Drug market.
What Is GDUFA Self-Identification?
The Generic Drug User Fee Amendments (GDUFA) require human generic Drug facilities, along with certain sites and organizations listed in a generic Drug submission, to submit identification information to the FDA every year. GDUFA self-identification is a mandatory process where these entities report their facility details and activities to the FDA for regulatory compliance.1
GDUFA was introduced in 2012 under the Food and Drug Administration Safety and Innovation Act (FDASIA) to improve the review process for generic Drugs and ensure timely access to safe and affordable medicines. It is renewed every five years, with GDUFA III currently in effect until 2027.2
Who Must Self-Identify Under GDUFA?
Generic Drug facilities, sites, and organizations must comply with FDA self-identification generic Drugs regulations. These entities include manufacturing, packaging, testing, and clinical sites involved in generic Drug compliance activities.
They must meet roles clearly specified outlined below:
- Facilities that manufacture or intend to manufacture generic APIs for human usage or finished dosage forms (FDFs).
- Packaging sites and organizations that place FDF into primary containers and do the labelling.
- Contract sites that remove products from primary containers and subdivide them into container systems.
- Bioequivalence and bioavailability testing sites conducting clinical, bioanalytical or in vitro studies for submissions.
- Contract testing sites performing CGMP-required analysis of dosage forms or active pharmaceutical ingredients.1
Information Required for GDUFA Self-Identification
To complete GDUFA self-identification, facilities must submit structured and standardized information to the FDA. This data helps ensure accurate tracking of sites involved in generic Drug development and manufacturing.
The required identification details include:
- Document information that defines the submission and supports electronic record management.
- Data Universal Numbering System (DUNS) number used to uniquely identify each entity.
- Registrant information covering the name, email address, phone and address, and DUNS of the registrant organization responsible for the facility or site.
- Facility information describing name, Facility DUNS number, FDA Establishment Identifier (FEI), contact information and location of the registrant.
- Type of business operations the registrant is involved in; e.g. API, FDF, etc. and whether non-generic Drugs are also manufactured at the same sites.
- Electronic submission data formatted in Structured Product Labelling (SPL) using XML standards through the ESG system.3
How to Submit GDUFA Self-Identification to the FDA
GDUFA self-identification must be submitted electronically using standardized formats approved by the FDA. The process ensures consistent data exchange and supports regulatory tracking across generic Drug facilities.
To complete submission, follow these key steps:
- Prepare self-identification data using the HL7 SPL format.
- Use one of the tools such as CDER Direct, eSubmitter, Xforms, or other compliant software to create, edit and submit SPL.
- Create and validate the SPL file according to FDA technical specifications.
- Obtain a WebTrader account for access to the Electronic Submissions Gateway (ESG).
- Submit the finalized SPL file through ESG to the FDA for processing.1
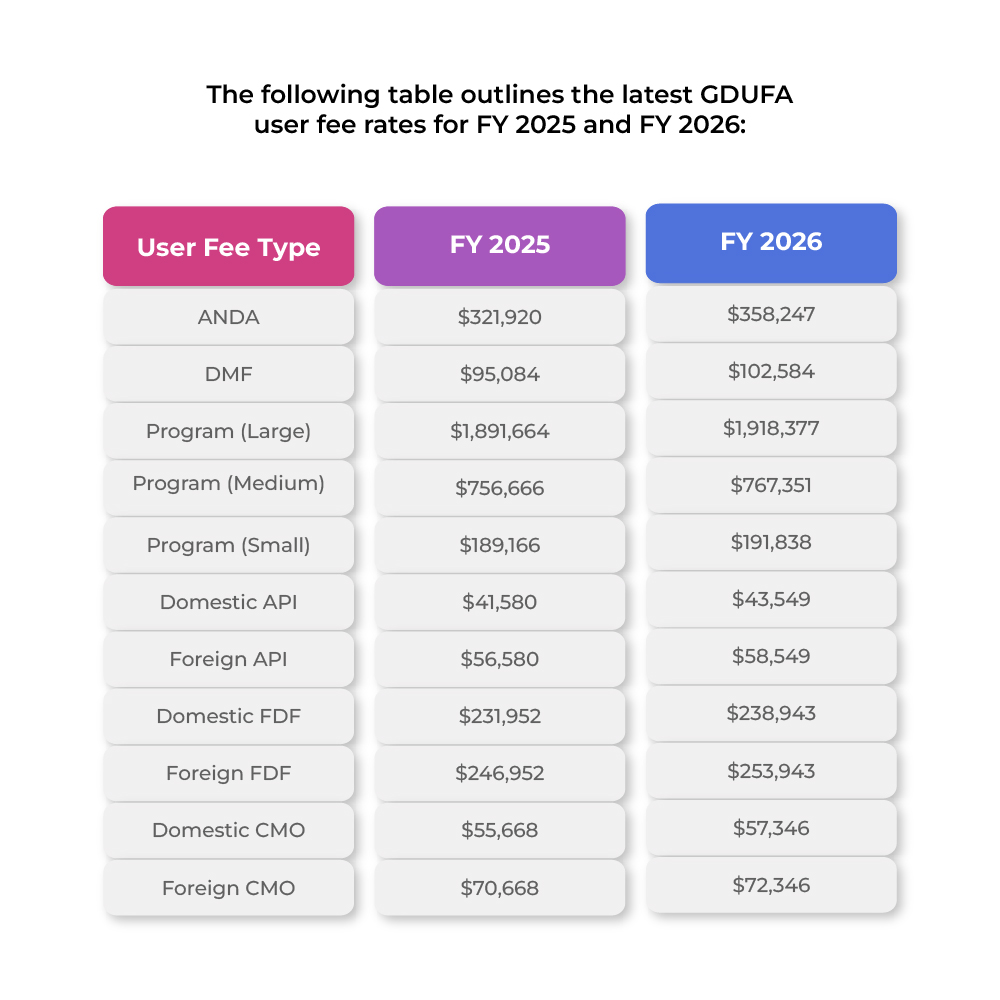
GDUFA Facility Fees and Payment Requirements
Under GDUFA, not all entities that complete self-identification are required to pay facility fees. Fee obligations apply mainly to facilities involved in manufacturing generic Drug components such as APIs (Active Pharmaceutical Ingredients) and FDFs (Finished Dosage Forms) to ensure FDA compliance generic Drugs.1 The program also operates under structured payment systems designed to improve efficiency and transparency in fee collection and regulatory processes.2
The key rules for determining fee responsibility are outlined below:1
- Only facilities that manufacture, or intend to manufacture, generic Drug APIs or FDFs are required to pay facility fees.
- Sites involved only in testing, repackaging, or relabelling are not required to pay any user fee.
- Facilities that package and/or label human finished dosage forms of generic Drugs are considered manufacturers and must pay annual FDF fees.
- This applies whether packaging is performed under contract or directly by the applicant.
- A facility is not required to pay more than one annual API fee and/or one annual FDF fee.
From October 1, 2025, the FDA no longer accepts paper-based payments, and all fees must be made electronically through methods such as Automated Clearing House (ACH), credit card, or wire transfer. The FDA also publishes monthly performance metrics and annual activity reports, improving transparency around submissions, approvals, and fee-related operations.2
FDA Penalties for GDUFA Non-Compliance
Under Generic Drug user fee amendments, facilities that do not complete required self-identification may affect the legal status of products manufactured at the facility. Failure to comply with self-identification requirements can result in the following outcomes:
- All FDF and API products from the facility are considered misbranded
- Shipping misbranded products within the United States is a violation of federal law
- Importing such products into the United States is also prohibited
- Enforcement actions may include prosecution, injunctions, or product seizures
- Products from non-compliant facilities may be denied entry into the United States1
Conclusion
GDUFA self-identification is an important FDA requirement for generic Drug facilities to ensure proper regulation and monitoring. It involves submitting accurate facility information, following standard formats, and meeting applicable fee obligations. Understanding who must comply and how to submit data helps avoid serious legal risks. Non-compliance can attract misbranding and import restrictions. Therefore, staying updated with GDUFA requirements (FDA) supports smooth approvals, regulatory compliance, and continued access to the US generic Drug market.
References
1. Self-Identification FAQs | FDA. Accessed April 9, 2026. https://www.fda.gov/industry/generic-drug-user-fee-amendments/self-identification-faqs
2. Research C for DE and. Generic Drug User Fee Amendments. FDA. April 7, 2026. Accessed April 9, 2026. https://www.fda.gov/industry/fda-user-fee-programs/generic-drug-user-fee-amendments
3. FDA. Generic Drug User Fee Amendment (GDUFA II) SPL Industry Technical Specification Information. Accessed April 9, 2026. https://www.fda.gov/media/84762/download
CliniExperts - Your reliable partner for Comprehensive Compliance Solutions. We offer 360 degree Global Regulatory Solutions related to Pharma, Medical Devices and In-Vitro Diagnostics.
CliniExperts
CliniExperts Services Pvt. Ltd.
Recent Posts
Risk Mitigation Through FDA Q-Submission Consulting

Short Description The FDA Q-Submission Program is an important regulatory pathway that helps Medical Device sponsors reduce potential risks before submitting a formal application. By obtaining early F..
What Is the FDA Q-Submission Program?

Short Description The FDA Q-Submission Program is a voluntary process that allows Medical Device developers to communicate with the United States Food and Drug Administration before submitting a forma..
Key Responsibilities of a Marketing Authorization Holder Under EU Regulations

A marketing authorization holder (MAH) plays a critical role in ensuring that medicinal products marketed in the European Union (EU) continue to meet Regulatory standards for quality, safety, and effi..
Contact us
Please feel free to talk to us if you have any questions. We endeavour to answer within 24 hours.