European Union Market
Clinical Evaluation of Medical Devices
Contact us today to learn how we can help improve your regulatory operations.

Medical Device


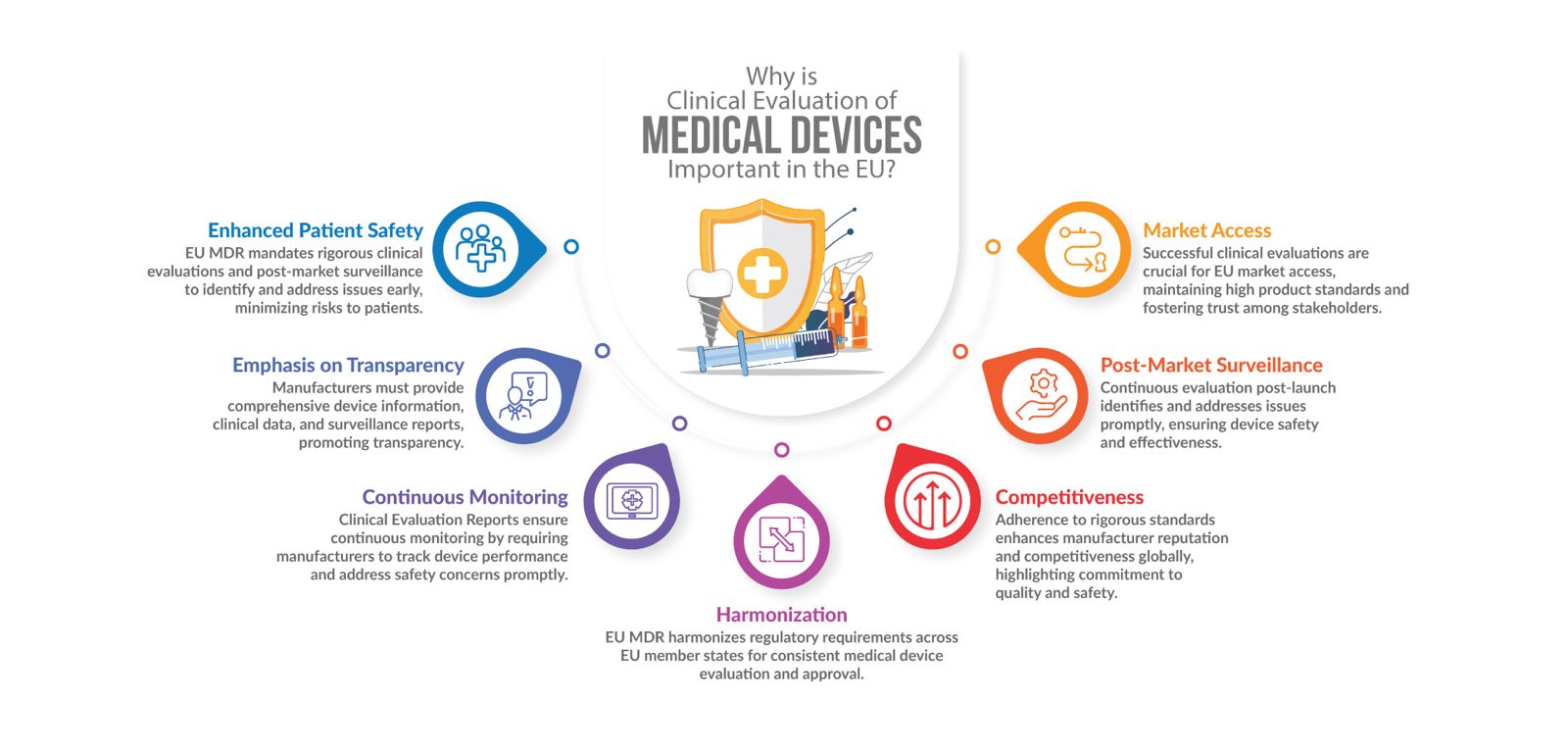
Why is Clinical Evaluation of Medical Devices Important in EU?
The new EU Regulation (EU) 2017/745 on medical devices, which came into force on May 26, 2017, is critical for medical device makers seeking CE certification and recertification of their devices. All medical devices to be sold in the European Union must comply with EU MDR requirements. On clinical evaluation, the current contribution analyses the significant changes between EU Directive 93/42/EEC and EU Regulation 2017/745 in the following six areas:
- Stronger requirements for clinical safety and evidence of clinical efficacy
- Classification
- Clinical evaluation, possibly including clinical trials
- Post-market clinical surveillance
- Clinical documentation and reporting, and
- Introduction of the European Commission’s scrutiny procedure
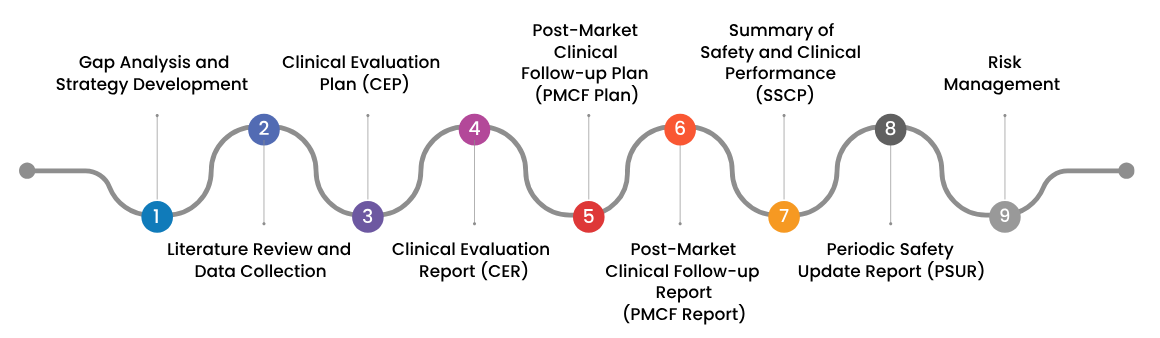
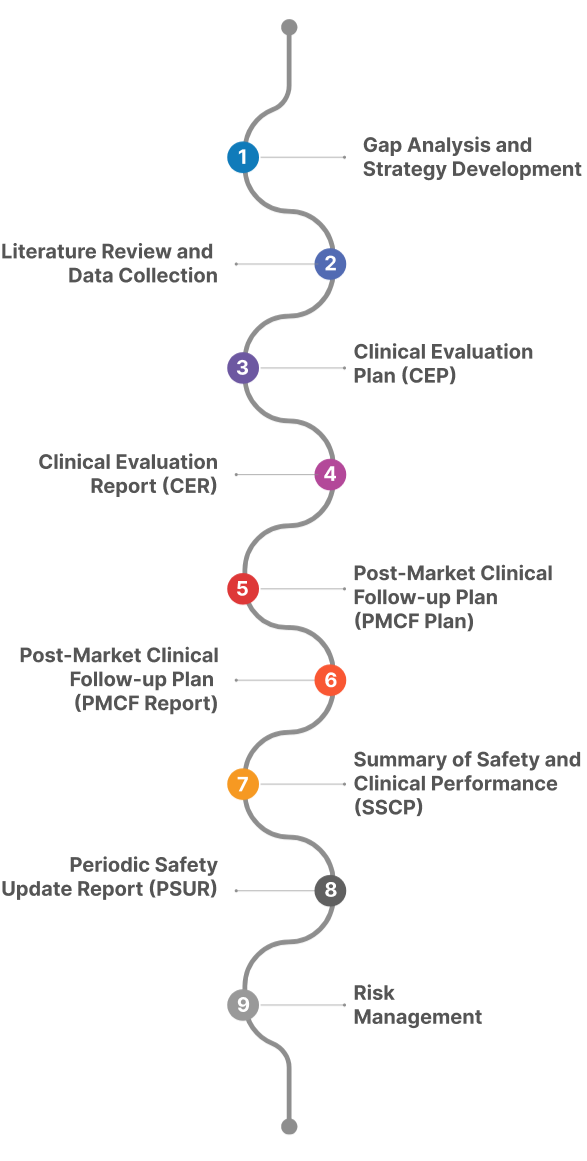
Process for conducting Clinical Evaluation of your Medical Devices:
The process of systematically and continuously evaluating clinical data to confirm the device’s performance and safety over its entire lifecycle is known as the clinical evaluation process for medical devices. The process of preparing a clinical evaluation report involves several key steps, including:
|
Identifying the regulatory requirements to be backed by the clinical information |
Identification of available clinical information for the medical devices under evaluation and state-of-the-art for proposed use. |
|
Determining the availability of clinical information for the safety and effectiveness of the devices under evaluation as well as it’s benefit-to-risk ratio. |
Generating additional clinical information if the available data is not sufficient. |
Scope and Clinical Evaluation Plan
Effective clinical evaluation requires careful planning. To prove that a device meets its intended performance, clinical benefits, and safety standards, it's crucial to first identify these parameters clearly.
MEDDEV 2.7.1 Rev 4 states that your first step should be to define the scope of the evaluation, which should include:
- The device description.
- Any design features, indications, or target populations that need specific attention during the evaluation.
- The information you need for evaluating equivalence, if you intend to claim it.
- Risk management documents such as the hazard identification list and any clinical risks identified from the risk analysis.
- Current knowledge applicable standards and guidance documents.
- The data sources and types you will use in the clinical evaluation.
Establishing equivalency with devices already on the market
Under EU MDR, the manufacturer should demonstrate that their device is equivalent in all three, (i) Clinical (ii) Technical, and (iii) Biological.
Identifying Relevant Data and Create a Literature Review Protocol
Once the scope for the clinical evaluation is established, a plan will be created to carry out. To identify any equivalent devices, the next step would be collecting relevant data.
Obtaining Clinical Data for the Clinical Evaluation
The clinical evaluation will be based on both direct and indirect data. Direct data is the one, which has been generated by the manufacturer’s device such as data obtained from the clinical trials or pilot studies. If the devices are Class IIb implantable or Class III, then the manufacturer will be required to conduct the clinical trial of their device. Direct data also includes publicly available data or registries on your device. The direct data can also be generated from the post-market surveillance activities.
Indirect clinical data is the one which has been generated by an equivalent device. The decision to claim equivalence (or not) will be one of the factors that affects your literature review.
Literature review
Literature review will have the two outputs: (i) Literature on the subject device and the equivalent device (ii) A review of the State-of-the-art which is necessary for appraisal and analysis of the clinical data from the literature on the subject device and equivalent device.
- You’ll need to designate specific terms that you’ll use in your search. If your terms are too broad, your search may return thousands of articles, many of which will be irrelevant to your clinical evaluation.
- Your literature search protocol also needs to establish which sources of information will be used for searches, and you need to be consistent about using those sources. That means applying the same search terms to several databases, such as PubMed, MEDLINE, or Embase.
MEDDEV 2.7.1 Rev 4 defines ‘state-of-the-art’ as: ‘Applicable standards and guidance documents, information relating to the medical condition managed with the device and its natural course, benchmark devices, other devices and medical alternatives available to the target population’.
Clinical data appraisal from the literature review
The following factors will be considered for appraising the clinical data: (i) suitability (ii) Applicability (iii) Population and, (iv) Quality.
Clinical Investigations
If the collected data related to safety or performance of the device does not provide strong evidence to determine the device’s compliance with GSPRs, in such case, manufacturer would need to generate the missing data to draw conclusions about the device’s conformity with GSPRS. This may require a clinical investigation.
Post-market clinical follow-up (PMCF) needs
The PMCF is a continuous process of updating medical device’s clinical evaluation once the manufacturer has obtained CE marking.
The final step in the data analysis stage is to describe any residual risks, uncertainties, or unanswered questions that your PCMF will need to collect data on once the device reaches market.
Clinical Evaluation Pathways under Article 61
Article 61 of the MDR outlines different pathways for clinical evaluation based on risk class and device novelty, which can be broadly categorized into:
- Clinical Investigations: Mandatory for most implantable and Class III Medical Devices, with a few exceptions
- Evaluation based on existing data: Leverages data from similar or identical devices for lower-risk devices
- Combined approach: Employs a combination of clinical investigations and existing data
How we can help with your Clinical Evaluation Process
We can help you in all the steps mentioned above by preparing all the documents that are required for the Clinical Evaluation of your device.