IVD Compliance In The EU: Essenti Manufacturers


Summary:
- Ensuring MDR compliance in the EU is crucial for manufacturers.
- Key steps include understanding MDR requirements, conducting gap analysis, developing a compliance strategy, updating technical documentation, implementing robust QMS, engaging with notified bodies, and maintaining continuous compliance.
- Partnering with EU medical device consulting experts like Cliniexperts can streamline this complex process.

Introduction
For manufacturers of in vitro diagnostic (IVD) devices seeking to market their products in the European Union, ensuring MDR compliance is a critical requirement. The EU Medical Device Regulation (MDR) has brought about significant changes in the regulatory landscape, and manufacturers must navigate these new rules to achieve and maintain compliance. Before divulging details, it is essential to understand IVD compliance.
Understanding IVD compliance
IVD (In Vitro Diagnostic) Compliance refers to the adherence of IVD devices to the regulatory requirements set forth by the European Union (EU) to ensure the safety and performance of these devices in the EU market.
The EU IVDR (In Vitro Diagnostic Medical Device Regulation) 2017/746, which came into effect on May 26, 2022, replaced the In Vitro Diagnostics Medical Device Directive 98/79/EC and introduced significant changes to the regulatory framework for IVD devices.
IVD devices are classified into four classes based on their intended purpose and inherent risk:
| Class of medical device | Risk of medical device |
| Class A | low individual and public health risks |
| Class B | moderate individual risks and potentially low public health risks |
| Class C | high individual risks and moderate public health risks |
| Class D | high individual risks and high public health risks |
Based on these classifications, manufacturers must apply for Medical device registration in EU with a proper justification. To ease the process for manufacturers, here is the step-by-step guide to understand IVDR and MDR compliance in detail.
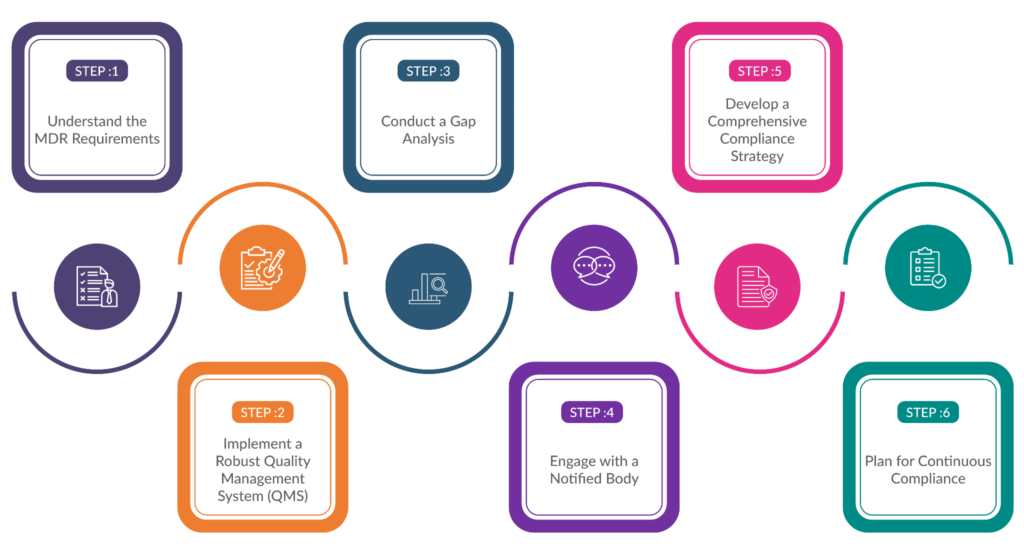
Step Involved in IVDR and MDR compliance
To ease the process for manufacturers, here is a step-by-step guide to understand IVDR and MDR compliance in detail.
Understand the MDR Requirements
The first step for manufacturers is to gain a comprehensive understanding of the MDR requirements. The regulation has introduced more stringent rules for clinical evidence, post-market surveillance, and vigilance reporting.
Additionally, it has expanded the scope of regulated devices, including certain products that were previously unregulated.
Manufacturers should consult reputable sources, such as the European Commission’s official website or EU medical device consulting firms like CliniExperts for guidance regarding documents from notified bodies, to ensure they have a thorough grasp of the applicable requirements.
Conduct a Gap Analysis
Once the MDR requirements are well understood, manufacturers should conduct a gap analysis to identify areas where their existing processes and documentation may not meet the new standards.
This analysis should cover various aspects, including technical documentation, clinical evaluation, risk management, and quality management systems.
Develop a Comprehensive Compliance Strategy
Based on the gap analysis, manufacturers should develop a comprehensive compliance strategy that outlines the steps and resources required to achieve MDR compliance.
This strategy should include timelines, budget allocations, and responsible personnel for each task.
It is important to prioritize high-risk areas and address any critical gaps first. Manufacturers may also consider seeking EU medical device consulting services from experienced professionals to ensure their compliance efforts are on the right track.
Update Technical Documentation
Manufacturers must update their technical documentation to meet the MDR requirements. This includes revising the clinical evaluation, risk management documentation, and instructions for use (IFUs).
The technical documentation should demonstrate how the device meets the essential safety and performance requirements outlined in the regulation.
Implement a Robust Quality Management System (QMS)
A robust QMS is essential for MDR compliance. Manufacturers should review and update their QMS to align with the new requirements, including those related to post-market surveillance, vigilance reporting, and complaint handling.
The QMS should also incorporate processes for monitoring and maintaining compliance on an ongoing basis, as well as mechanisms for addressing any non-conformities or corrective actions.
Engage with a Notified Body
Under the MDR, most IVD devices require intervention from a notified body, which is an independent organization designated by the European Commission to assess and certify medical devices.
Manufacturers should select a notified body and initiate the conformity assessment process early on.
This process typically involves submitting technical documentation, undergoing audits, and obtaining the necessary certifications or approvals from the notified body.
Plan for Continuous Compliance
MDR compliance is an ongoing process, and manufacturers must be prepared to maintain compliance throughout the product lifecycle.
This includes monitoring and addressing any changes in regulatory requirements, updating technical documentation as needed, and implementing effective post-market surveillance and vigilance reporting processes.
Manufacturers should also plan for periodic audits and assessments by the notified body to ensure continued compliance and maintain their certifications.
By following these essential steps, manufacturers of IVD devices can navigate the complexities of the EU medical device registration process and achieve MDR compliance.
Failure to comply with the MDR can result in significant consequences, including product recalls, fines, and potential market exclusion.
Manufacturers are encouraged to seek guidance from regulatory experts, industry associations, and EU medical device consulting firms to ensure they are taking the appropriate measures to meet the MDR requirements and maintain compliance throughout the product lifecycle.
CliniExperts, a leading EU medical device consulting firm, offers comprehensive services to help IVD manufacturers achieve and maintain MDR compliance.
With a team of experts well-versed in the intricacies of the EU regulatory landscape, CliniExperts provides tailored solutions, including gap analysis, technical documentation review, quality management system implementation, and guidance throughout the medical device registration EU process.
Their end-to-end support ensures manufacturers navigate the complexities of the MDR seamlessly, mitigating compliance risks and facilitating successful market access in the European Union.
Conclusion
In conclusion, navigating the intricate world of IVD compliance in the EU requires a strategic approach and meticulous attention to detail. By following the essential steps outlined and leveraging the expertise of EU medical device consulting firms like CliniExperts, manufacturers can confidently achieve and maintain MDR compliance, enabling them to market their products successfully in the European market.
References
- European Commission. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. [Internet]. [cited 2024 Apr 24]. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32017R0746
- European Commission. MDR: Medical Devices Regulation. [Internet]. [cited 2024 Apr 24]. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=LEGISSUM%3A4301047&qid=1713967419255
- European Commission. Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices. [Internet]. [cited 2024 Apr 24].Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
- Medical Device Regulation. (2019). Step-by-Step Guide to MDR. [Internet]. [cited 2024 Apr 24]. Available from: https://www.medical-device-regulation.eu/wp-content/uploads/2019/06/md_mfr_stepbystep.pdf
- Medical Device Regulation. (2020). White Paper: Technical Documentation. [Internet]. [cited 2024 Apr 24]. Available from: https://www.medical-device-regulation.eu/wp-content/uploads/2020/09/White_Paper__Technical_Documentation_Web_v3.pdf
CliniExperts - Your reliable partner for Comprehensive Compliance Solutions. We offer 360 degree Global Regulatory Solutions related to Pharma, Medical Devices and In-Vitro Diagnostics.
CliniExperts
CliniExperts Services Pvt. Ltd.
Recent Posts
Risk Mitigation Through FDA Q-Submission Consulting

Short Description The FDA Q-Submission Program is an important regulatory pathway that helps Medical Device sponsors reduce potential risks before submitting a formal application. By obtaining early F..
What Is the FDA Q-Submission Program?

Short Description The FDA Q-Submission Program is a voluntary process that allows Medical Device developers to communicate with the United States Food and Drug Administration before submitting a forma..
Key Responsibilities of a Marketing Authorization Holder Under EU Regulations

A marketing authorization holder (MAH) plays a critical role in ensuring that medicinal products marketed in the European Union (EU) continue to meet Regulatory standards for quality, safety, and effi..
Contact us
Please feel free to talk to us if you have any questions. We endeavour to answer within 24 hours.