The Importance of Comprehensive Documentation for MDR Compliance


Summary:
Comprehensive documentation is crucial for MDR compliance, enabling transparency, traceability, and accountability throughout the medical device lifecycle. Rigorous documentation of design, risk management, clinical evaluations, manufacturing, and post-market surveillance demonstrates adherence to safety and performance requirements, facilitating regulatory approval and public trust.

The Significance of Thorough Documentation for MDR Compliance
Introduction
On 26 May 2017, the Medical Device Regulation (MDR) entered into force, replacing the previous Medical Devices Directive (MDD). By 26 May 2020, manufacturers needed to ensure that their technical documentation complied with the MDR Compliance to obtain or renew a CE certificate or issue a Declaration of Conformity (DoC).
The European Union’s Medical Device Regulation (MDR) (2017/745) has fundamentally reshape the landscape for placing medical devices on the market within the European Economic Area (EEA). The MDR, which replaced Directives 93/42/EEC (MDD) and 90/385/EEC (AIMDD), emphasizes a stricter approach to ensuring the safety, efficacy, and performance of these vital tools.2 However, amidst the focus on rigorous testing and risk management, one crucial element often takes centre stage: documentation.
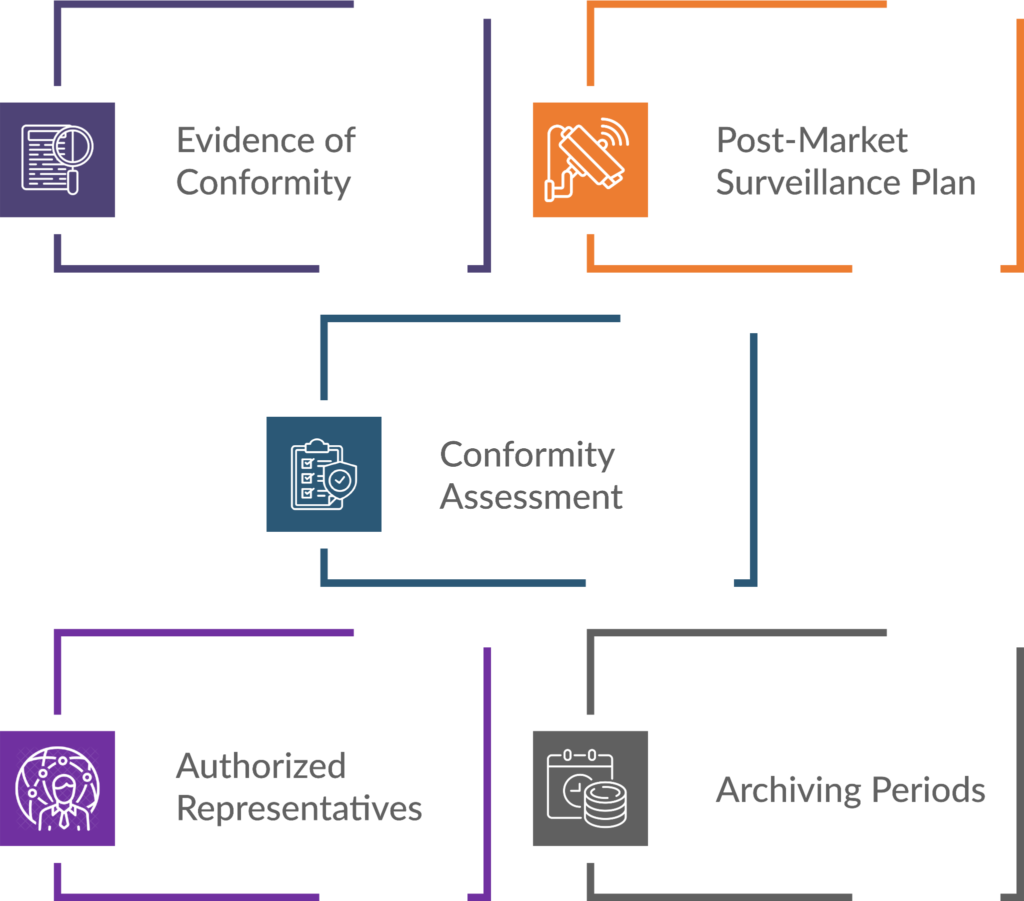
Key Elements of Technical Documentation for MDR Compliance
- Evidence of Conformity: Technical documentation provides evidence that a medical device complies with safety and performance requirements. It includes details about the device’s design, manufacturing processes, risk assessments, and clinical evaluations.
- Post-Market Surveillance Plan: Manufacturers must outline how to monitor the device’s performance once it’s on the market. This plan ensures ongoing safety and effectiveness.
- Conformity Assessment: The MDR mandates a thorough technical documentation review by Competent Authorities (CAs) and Notified Bodies (NBs). Clear and well-organised documentation facilitates this assessment.
- Authorised Representatives: Manufacturers must designate an authorised representative within the EU. Documentation should include information about this representative.
- Archiving Periods: Manufacturers need to retain technical documentation for specified periods. Proper archiving ensures traceability and accountability.
MDR Compliance: A Documentation-Driven Process
The MDR adopts a risk-based classification system for medical devices. This means the level of documentation required will vary depending on your device’s classification (Class I, Class IIa, Class IIb, or Class III). However, regardless of classification, a robust documentation system remains the cornerstone of a successful MDR compliance strategy.
Manufacturers are mandated to establish and maintain a documented Quality Management System (QMS). This system should encompass documented procedures, processes, and records for the device’s entire lifecycle, spanning design and development, manufacturing, post-market surveillance, and potential corrective actions.
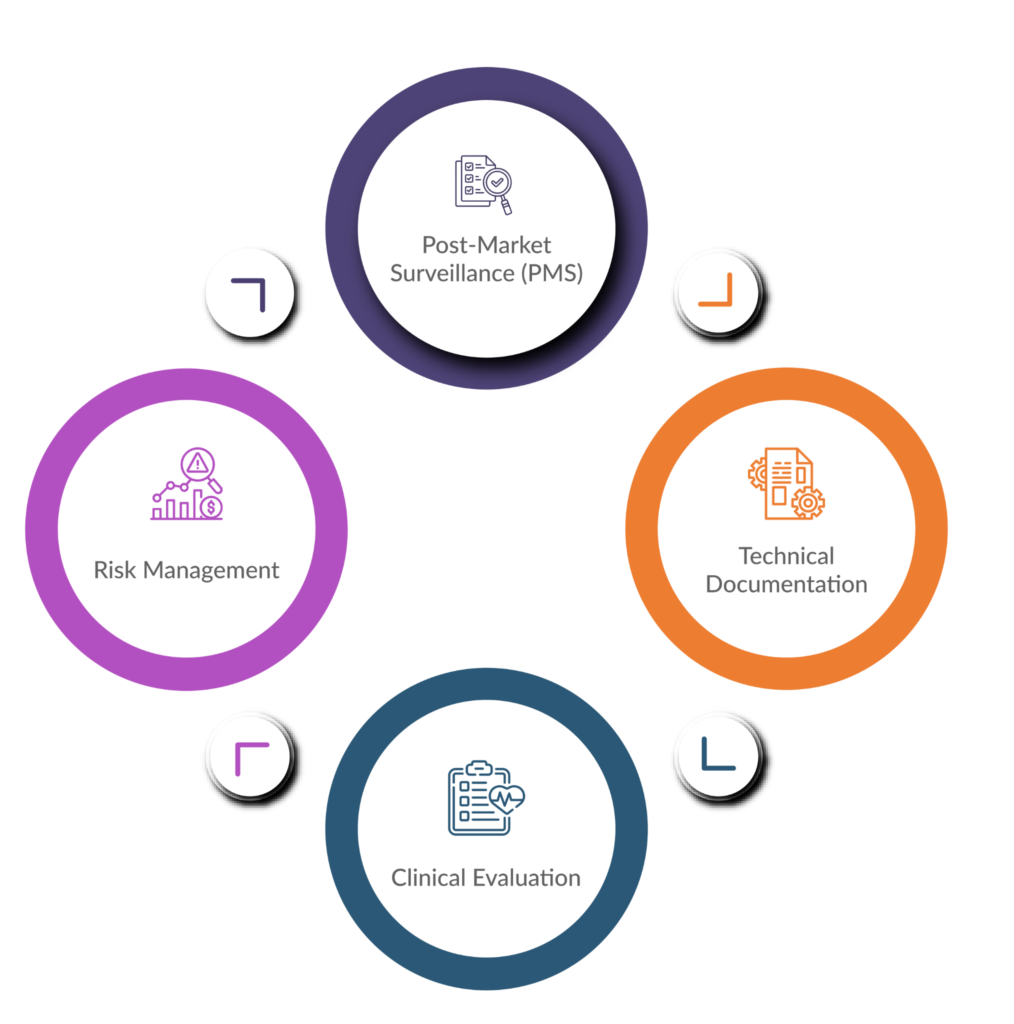
Here’s a closer look at key areas where comprehensive documentation plays a vital role in MDR compliance:
Technical Documentation
This forms the foundation of your application for a CE marking certificate. It details the design and development specifications, risk management procedures, performance evaluation data, and labelling information for your device.
Risk Management
The MDR necessitates a robust risk management process. You’ll need documented risk analyses that identify potential hazards, assess their severity and likelihood, and outline mitigation strategies. A 2020 article published in the International Journal of Medical Engineering & Informatics emphasizes the importance of well-documented risk management activities for ensuring patient safety.
Clinical Evaluation
For higher-risk devices, clinical evaluation plans and reports demonstrating the device’s safety and performance become necessary. This documentation should adhere to the MDR’s specific requirements for clinical investigations, as outlined on the European Commission website.
Post-Market Surveillance (PMS)
The MDR emphasizes ongoing monitoring of your device’s performance once it’s on the market. This necessitates a documented PMS plan outlining how you’ll collect and analyse data on adverse events, incidents, and corrective actions.
The Advantages of Comprehensive Documentation
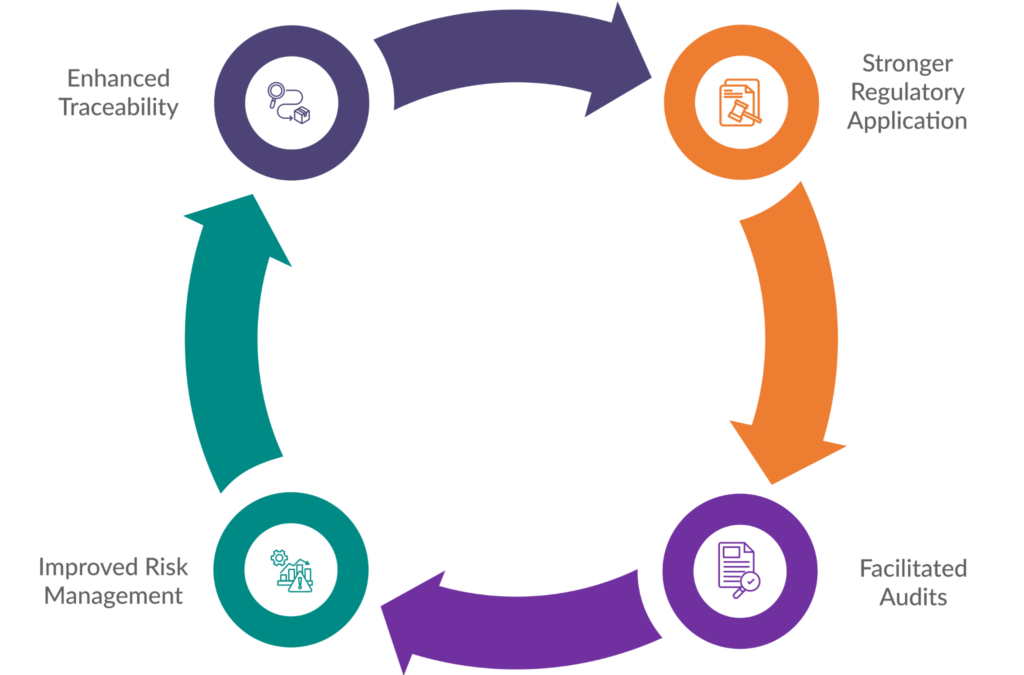
Maintaining meticulous documentation offers significant advantages for medical device manufacturers seeking EU market access:
Stronger Regulatory Application
A well-organized and complete set of technical documentation demonstrates to Notified Bodies (NBs) that you understand the medical device registration EU requirements and have implemented a robust QMS. This strengthens your application for a CE marking certificate and expedites the approval process.
Facilitated Audits
Regular audits by NBs are a reality under MDR. Comprehensive documentation ensures you have all the necessary records readily available, allowing for a smoother and less stressful audit experience.
Improved Risk Management
Detailed documentation of your risk management activities fosters a proactive approach to safety. It enables you to identify and address potential risks early on, minimizing the likelihood of post-market issues.
Enhanced Traceability
Thorough documentation facilitates traceability throughout the device’s lifecycle. This allows you to track components, materials, and production processes effectively, ensuring quality control and facilitating swift corrective actions if needed.
As a leading provider of EU medical device consulting services, CliniExperts understands the paramount importance of comprehensive documentation in achieving and maintaining MDR compliance. Our team of experts offers tailored solutions to guide medical device manufacturers through the intricate documentation requirements of the MDR, ensuring transparency, traceability, and adherence to the highest standards of safety and quality.
Conclusion
In summary, technical documentation is not just paperwork for medical device registration EU—it’s a critical component of MDR compliance. Properly documented evidence ensures that medical devices meet safety and performance standards. Manufacturers, regulatory authorities, and patients all benefit from well-organized and transparent documentation.
References
- Medical-device-regulation. [Internet]. [cited 2024 Apr 24]. Available from: https://www.medical-device-regulation.eu/wp-content/uploads/2020/09/White_Paper__Technical_Documentation_Web_v3.pdf
- Ensuring the safety and performance of medical devices. [Internet]. [cited 2024 Apr 24]. Available from: https://eur-lex.europa.eu/EN/legal-content/summary/ensuring-the-safety-and-performance-of-medical-devices.html
- Lex – 12016P/TXT – EN – EUR-Lex. [Internet]. [cited 2024 Apr 24]. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
CliniExperts - Your reliable partner for Comprehensive Compliance Solutions. We offer 360 degree Global Regulatory Solutions related to Pharma, Medical Devices and In-Vitro Diagnostics.
CliniExperts
CliniExperts Services Pvt. Ltd.
Recent Posts
Risk Mitigation Through FDA Q-Submission Consulting

Short Description The FDA Q-Submission Program is an important regulatory pathway that helps Medical Device sponsors reduce potential risks before submitting a formal application. By obtaining early F..
What Is the FDA Q-Submission Program?

Short Description The FDA Q-Submission Program is a voluntary process that allows Medical Device developers to communicate with the United States Food and Drug Administration before submitting a forma..
Key Responsibilities of a Marketing Authorization Holder Under EU Regulations

A marketing authorization holder (MAH) plays a critical role in ensuring that medicinal products marketed in the European Union (EU) continue to meet Regulatory standards for quality, safety, and effi..
Contact us
Please feel free to talk to us if you have any questions. We endeavour to answer within 24 hours.