Marketing Authorization Holders in the EU and the Regulatory Requirements


Summary:
The regulatory environment for in the European Union is a complex framework designed to uphold the safety, efficacy, and quality of medicinal products. Let’s explore how MAHs navigate rigorous compliance requirements set forth by the European Medicines Agency and notified bodies to ensure market approval, safeguarding public health through meticulous adherence to EU regulations.

Introduction
Marketing Authorization Holders (MAHs) in the European Union (EU) play a crucial role in ensuring the safety, efficacy, and quality of medicinal products and medical devices.
The regulatory environment in the EU is structured to safeguard public health by enforcing stringent compliance standards for MAHs.
Understanding these regulations is essential for MAHs to maintain market authorization and ensure their products meet the high standards set by the EU.
What is a Marketing Authorization Holder (MAH)?
MAH in the EU is responsible for securing and maintaining the marketing authorization for medicinal products and medical devices.
This authorization, granted by competent authorities or the European Commission, ensures that products meet rigorous standards of safety, efficacy, and quality before entering the EU market.
MAHs play a critical role in overseeing the regulatory compliance and market entry of these healthcare products.2
Regulatory Framework and Authorities
The regulatory framework for MAHs in the EU encompasses various authorities and stringent standards:
European Medicines Agency (EMA):–
The EMA oversees the scientific evaluation, and safety monitoring of medicinal products and medical devices across the EU.
It provides scientific opinions and recommendations to support decision-making of national competent authorities and the European Commission.
Notified Bodies:–
Designated by EU member states, notified bodies assess the conformity of medical devices with EU regulations.
They conduct thorough conformity assessments, issue CE marks for compliance, and may consult expert panels for scientific opinions on high-risk devices.
European Commission:
The European Commission grants marketing authorizations based on evaluations from the EMA and national competent authorities.
It ensures that authorized medicinal products and medical devices meet EU-wide standards before market entry.
In essence, this regulatory framework and collaboration among these authorities are crucial in safeguarding public health by ensuring that products meet stringent criteria for safety, efficacy, and quality in the European Union.
Steps to Obtain EU Marketing Authorization
Securing EU marketing authorization involves a series of structured steps to ensure compliance and approval across member states.
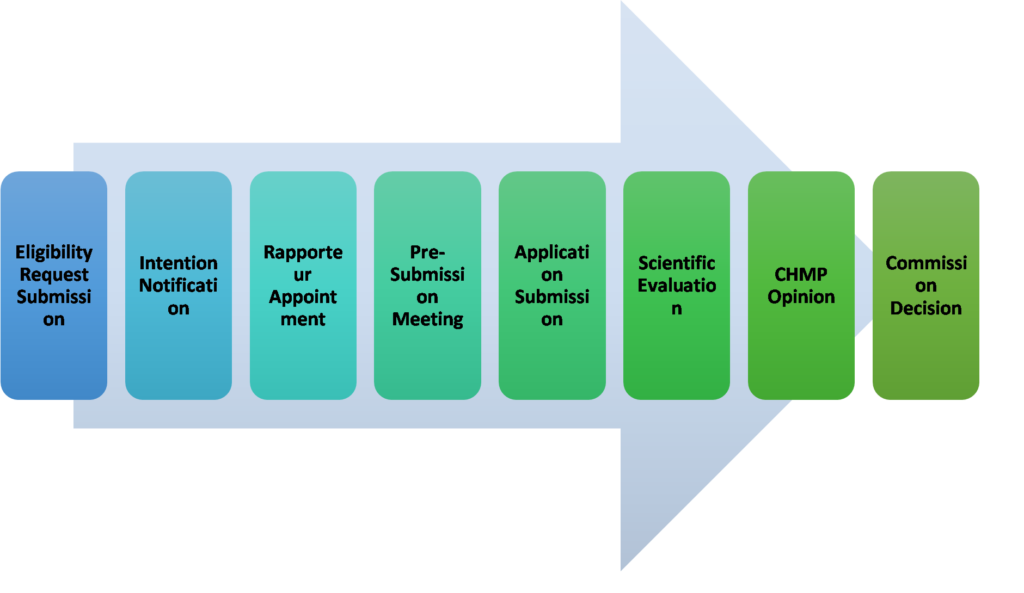
Figure 1: Steps to Obtain EU Marketing Authorization
Eligibility Request Submission:
MAHs initiate the process by submitting an eligibility request 18 to 7 months prior to the planned application submission.
This step determines if the product qualifies for the centralized procedure, which facilitates approval across EU member states.
Intention Notification:
Approximately 7 months before submitting the application, MAHs notify regulatory authorities of their intention.
This notification initiates early discussions with authorities and allows for initial preparations regarding regulatory requirements.
Rapporteur Appointment:
Early in the process, MAHs appoint rapporteurs or co-rapporteurs. These experts guide the evaluation process, ensuring thorough assessment of the product’s quality, safety, and efficacy according to EU standards.
Pre-Submission Meeting:
Held approximately 7 months before submission, this meeting is crucial for discussing specific details of the application with regulatory authorities.
It helps clarify expectations and requirements, ensuring a smoother submission process.
Application Submission:
MAHs formally submit their application for marketing authorization to regulatory authorities.
This submission includes comprehensive data on the product’s safety, efficacy, and quality, obtained through rigorous testing and clinical trials.
Scientific Evaluation:
Regulatory authorities conduct a thorough scientific evaluation over a 210-day period.
This evaluation assesses the product’s compliance with EU regulations and its suitability for market approval based on scientific data and evidence.
CHMP Opinion:
The Committee for Medicinal Products for Human Use (CHMP) provides a scientific opinion based on the evaluation findings.
This opinion guides the European Commission’s final decision on granting or denying marketing authorization.
Commission Decision:
Based on the CHMP’s opinion and evaluation outcomes, the European Commission reviews the application.
It makes a decision to grant or deny marketing authorization, ensuring the product meets stringent EU standards for safety, efficacy, and quality.
Upon completing these structured steps, the European Commission carefully evaluates the application to verify compliance with stringent EU standards for safety, efficacy, and quality, culminating in the decision to grant or deny marketing authorization.
Responsibilities of a Marketing Authorization Holder (MAH)
According to EU guidelines, the responsibilities of a Marketing Authorization Holder (MAH) are extensive and crucial to ensure the safety and effectiveness of products in the market. These include:
Ensuring Regulatory Compliance:
MAHs uphold stringent EU regulations to ensure the accuracy and reliability of data supporting veterinary medicines, including coordinating inspections and analysing samples.
Overseeing Clinical Trial Execution:
MAHs oversee the planning and implementation of clinical trials, ensuring adherence to EU standards and ethical guidelines to collect crucial safety and efficacy data.
Managing Pharmacovigilance Activities:
MAHs compile clinical trial data, develop comprehensive risk management plans, and oversee long-term safety monitoring through patient registries, ensuring proactive management of adverse events.
Developing Robust Risk Management Strategies:
MAHs craft and update detailed Risk Management Plans (RMPs), delineating safety profiles and strategies to mitigate risks, adapting to emerging safety data and evolving regulatory demands.7
Navigating Changes in Device Integration:
MAHs monitor and manage modifications to devices, adhering to regulatory requirements for submitting variations, evaluating device performance, and conducting usability studies to uphold product safety and efficacy.
The MAH carries the ultimate responsibility to guarantee that products placed on the market meet stringent safety and efficacy standards as required by EU regulations.
Emerging Trends and Challenges
The regulatory landscape for MAHs in the EU continues to evolve in response to emerging trends and challenges:
Digital Health Innovations:
Advances in digital health technologies, including software as medical devices (SaMD), pose new regulatory challenges.
The EMA and EU authorities are adapting frameworks to ensure safety, efficacy, cybersecurity, and data privacy in these innovations.
Global Regulatory Harmonization:
Efforts to harmonize regulatory requirements globally aim to facilitate market access for MAHs across different jurisdictions while maintaining high standards of patient safety and product quality.
This ongoing evolution underscores the need for MAHs to navigate and adapt to these advancements to maintain compliance and meet market demands effectively.
Conclusion
The regulatory environment for Marketing Authorization Holders in the EU is designed to protect public health by enforcing rigorous standards for medicinal products.
MAHs must navigate a complex framework of regulations and guidelines, ensuring compliance in areas such as pharmacovigilance, regulatory compliance, quality management, and product information.
By fulfilling these responsibilities, MAHs play a vital role in maintaining the safety and efficacy of medicinal products available in the EU market.
Summary
- The European Union regulatory framework, led by the European Medicines Agency and notified bodies overseen by the European Commission, ensures the safety, efficacy, and quality of medicinal products.
- Obtaining European Union marketing authorization involves structured steps, including eligibility requests, Committee for Medicinal Products for Human Use evaluation, and thorough compliance across member states.
- Marketing authorization holders manage critical responsibilities such as clinical trials, good manufacturing practice-compliant manufacturing, complaints and recalls, pharmacovigilance, and supply chain oversight.
- Emerging digital health technologies, like software as a medical device, present regulatory challenges concerning safety, efficacy, cybersecurity, and data privacy.
- Global efforts to harmonize regulatory requirements aim to enhance market access while maintaining high standards of patient safety and product quality.
References
- Official Address Domenico Scarlattilaan 6 • 1083 HS Amsterdam • the Netherlands [Internet]. Available from: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/questions-answers-implementation-medical-devices-vitro-diagnostic-medical-devices-regulations-eu-2017-745-eu-2017-746_en.pdf
- Committee for Medicinal Products for Human Use (CHMP) Guideline on Quality Documentation for Medicinal Products When Used with a Medical Device Draft Agreed by Quality Working Party, Biologics Working Party and Guideline on Quality Documentation for Medicinal Products When Used with a Medical Device [Internet]. 2021. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-documentation-medicinal-products-when-used-medical-device-first-version_en.pdf
- Medical Devices | European Medicines Agency [Internet]. www.ema.europa.eu. Available from: https://www.ema.europa.eu/en/human-regulatory-overview/medical-devices#related-content-19159
- An Agency of the European Union Applying for EU Marketing Authorisation for Medicinal Products for Human Use [Internet]. Available from: https://www.ema.europa.eu/en/documents/leaflet/applying-european-union-marketing-authorisation-medicinal-products-human-use_en.pdf
- Compliance: Marketing Authorisation | European Medicines Agency [Internet]. www.ema.europa.eu. Available from: https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/compliance-marketing-authorisation
- Pharmacovigilance: Overview | European Medicines Agency [Internet]. www.ema.europa.eu. Available from: https://www.ema.europa.eu/en/human-regulatory-overview/pharmacovigilance-overview
- Risk Management | European Medicines Agency [Internet]. www.ema.europa.eu. Available from: https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/pharmacovigilance-marketing-authorisation/risk-management
CliniExperts - Your reliable partner for Comprehensive Compliance Solutions. We offer 360 degree Global Regulatory Solutions related to Pharma, Medical Devices and In-Vitro Diagnostics.
CliniExperts
CliniExperts Services Pvt. Ltd.
Recent Posts
Global Regulatory Strategy for Medical Devices – A CliniExperts Guide

This Article is all about Global Regulatory Strategy for Medical Devices – A CliniExperts Guide Introduction Medical Device manufacturers who are planning to enter the global market to market ..
21 CFR 801 vs. 21 CFR 803: Understanding Labelling and Reporting Regulations

Summary Short Description The 21 CFR 801 and 21 CFR 803 provide medical device regulations and cover labelling and Medical Device Reporting (MDR) requirements, respectively. What Is 21 CFR 801? 21 CFR..
The Role of US FDA 510(k) Consultants in Medical Device and IVD Approvals

Summary Short Description Navigating the U.S. Food and Drug Administration’s (FDA) 510(k) premarket notification process is a critical step for medical device and in vitro diagnostic (IVD) manuf..
Contact us
Please feel free to talk to us if you have any questions. We endeavour to answer within 24 hours.