Post-Market Surveillance (PMS) for Medical Devices


Summary:
- Post Market Surveillance or PMS is a regulatory requirement.
- It is a series of activities that monitors medical devices after being launched in the market for continued safety and efficacy.
- PMS is an important part of the pharmacovigilance of a medical device’s lifecycle.
- PMS helps collect safety information and evaluate the efficacy and performance of medical devices.
- PMS helps differentiate faulty devices from those that are safe and efficient.
- It helps identify and mitigate previous unrecognized adverse reactions and change patterns of adverse effects.
- PMS for medical devices allows for collecting quality, safety, and performance data throughout a device’s lifetime and helps manufacturers to build a complete risk-benefit profile for their devices.

(PMS) Post-Market Surveillance for medical devices is a regulatory requirement. It is mandatory in global markets including the United States and European Union.
PMS comprises of activities that relies on data gathering. It helps collect safety information and evaluate the efficacy and performance of medical devices that are already on the market.
PMS enables to report adverse events from a device post-market. PMS is designed to differentiate faulty devices from those that are safe and efficient for human use.
(PMS) Post-Market Surveillance For Medical Devices
Post-market surveillance, or PMS, to understand in simpler terms, refers to a series of activities typically conducted by manufacturers to monitor the safety, efficacy and performance of medical devices that have been launched on the market after the successful completion of clinical trials.
PMS monitoring helps to identify the need to take any precautionary action. It enables the manufacturers to screen their medical devices after being approved for sale and in-market use.
Post-Market Surveillance is a regulatory requirement in global markets which includes the United States and European Union.
Objectives of Post Market Surveillance
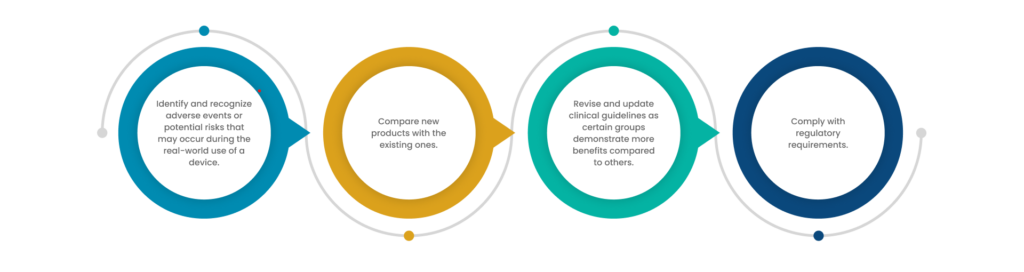
- The main objective of conducting PMS is to identify previously unrecognized adverse events and other additional positive effects of a particular medical device.
- PMS allows monitoring of medical devices in larger population groups to gather more data and information than that is collected during a clinical trial phase.
Evolution of Post Market Surveillance
Over the past few years, PMS practices have undergone changes with regulatory authorities after realizing the importance of applying measures to reduce the increasing adverse events.
PMS has been engineered as a proactive approach rather than a reactive approach. It has been used to focus on risk prevention and obligatory communication measures. In the global market, the United Kingdom (UK) and Canada are two of the most regulated pharmaceutical markets which have engaged in PMS.
Post Market Surveillance in the United Kingdom
Any medical device adverse events evident by the manufacturers should be directly reported to the Regulatory bodies such as FDA in the United States, MHRA in the United Kingdom, Health Canada in Canada etc. And also, since 2021, an additional requirement is needed to receive a UKCA mark as well.
Pharmacovigilance in the UK is practiced in the form of the Yellow Card Scheme. It is jointly operated by the Medicines and Healthcare Products Regulatory Agency (MHRA) and the Committee of Human Medicines (CHM).
Yellow Card Scheme is one of the first pharmacovigilance schemes. It comprises of the following objectives-
- It aims to identify and mitigate unrecognized adverse reactions and also change patterns of adverse effects.
- It monitors the use of medicines in everyday practice.
- It carries out risk–benefit analysis for medicines and suggests suitable actions, if and when necessary.
- It provides regular updates to healthcare professionals and patients with regard to the safety and efficacious use of medicines.
Importance of PMS for Medical Devices
Post-market surveillance for medical devices is critical for a device’s lifecycle. It is mandatory for manufacturers to investigate each medical device and inform the regulatory authorities within a specified time to provide their assessment related to the benefit/risk ratio as well as its safety and effectiveness in the post-market phase.
Medical device manufacturers can use real-life evidence from PMS-
- to detect adverse events as part of pharmacovigilance
- to conform with regulatory requirements
- to compare a new device’s performance against existing standards of care
- to continue monitoring the safety and effectiveness of the device in the intended patient population.
Manufacturers can collect the data on related devices from competitors as a part of PMS. This data can be collected as an observational analysis such as-
- internal observation done by the manufacturer’s internal databases that are collected as a post-market customer complaint, failure analysis and/or adverse event report. Such internal observations data is compared to the sales figure of the devices to continue to assess its risk-benefit ratio.
- external observation done by country specific regulatory authorities having medical device databases.
Steps to Conduct Post-Market Surveillance
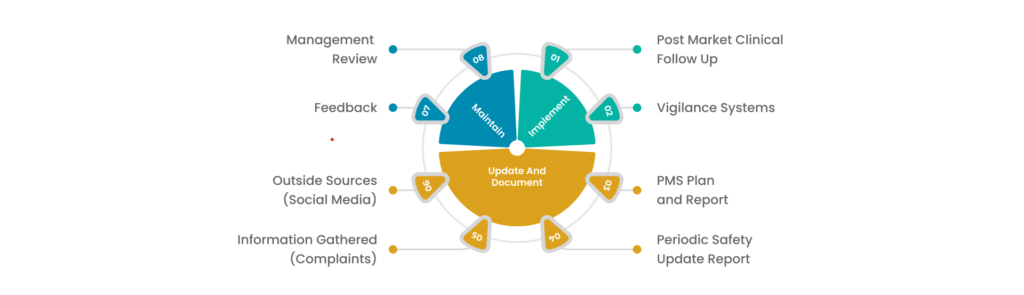
Usually, manufacturers of medical devices conduct PMS. At high levels, manufacturers may take the following steps to carry out PMS for their medical device vigilance-
- Step 1- The manufacturer must develop a PMS plan. It should include an assessment of whether Post-Market Clinical Follow-up (PMCF) is required or not.
- Step 2- The manufacturer must implement the assessment plan.
- Step 3- The manufacturer must generate PMS reports based on the findings.
PMS plan must comprise monitoring and data collection strategies. It must enable manufacturers to gather safety information on the medical devices.
PMS plan can be an integral part of the devices’ technical documentation which outlines the criteria for the risk-benefit assessment of the device.
PMS plan must include-
- approach for collecting and analyzing data
- proposal for following up on collected complaints
- communication of information to regulators and users
- strategies for taking corrective actions on devices
Post-market surveillance in the Different Countries
Based on different regions, manufacturers must comply with PMS regulations where they intend to sell their medical devices. In the US, PMS is only needed for higher-risk medical devices.
- The FDA uses Med Watch for consumers and healthcare experts to submit adverse event reports.
- 21 CFR Part 822 details are mandatory for PMS in the US.
- FDA uses the MAUDE database to submit medical device reports provided to the FDA by mandatory reporters.
Requirements for the European Union PMS
In the EU, PMS is mandatory for all medical devices. Manufacturers planning for a sale in the EU must prove that the medical device has performed Post-Market Clinical Follow-Up (PMCF) plan for their medical devices.
PMS has become a mandatory surveillance based on one single core principle that patient health and safety are critical factors that need to be considered when manufacturing and marketing medical devices.
Implementing PMS may fulfil post-approval needs of assessing and monitoring the risks accompanied by the use of medical devices in a larger population. It also enables spontaneous reporting of adverse reactions caused due to faulty devices. PMS safeguards the critical lives of patients.
Reference:
- Raj N, Fernandes S, Charyulu NR, Dubey A, S. RG, Hebbar S. Postmarket surveillance: a review on key aspects and measures on the effective functioning in the context of the United Kingdom and Canada. Ther Adv Drug Saf [Internet]. 2019 Jan [cited 2023 Jul 2]; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6661791/.
- Guidance for post-market surveillance and market surveillance of medical devices, including in vitro diagnostics [Internet]. [cited 2023 Jul 1]. Available from: https://www.who.int/publications/i/item/9789240015319
- Achieve EU MDR and UKCA medical device compliance – Free Guide 2022 [Internet]. [cited 2023 Jun 25]. Available from: https://www.mantrasystems.co.uk/eu-mdr-compliance/
CliniExperts - Your reliable partner for Comprehensive Compliance Solutions. We offer 360 degree regulatory solutions to Medical Devices and In-Vitro Diagnostics.
International CliniExperts
CliniExperts Services Pvt. Ltd.
Recent Posts
Step-by-Step Guide to ISO 13485 Certification in the UK

In the dynamic world of medical devices, adhering to stringent quality standards is paramount. ISO 13485 stands out as a beacon of excellence, setting forth requirements for a robust quality managemen..
Common Challenges in Medical Devices QMS Audits and How to Overcome Them

Implementing EU Quality Management Services is vital for medical device manufacturers to meet regulatory standards. Common challenges faced during QMS audits include internal audit gaps, leadership is..
The Benefits of Achieving ISO 13485 Certification for Your Medical Device Business in the UK

ISO 13485 certification is essential for UK medical device businesses, ensuring compliance with international standards and UK quality management certification requirements. It covers the entire produ..
Contact us
Please feel free to talk to us if you have any questions. We endeavour to answer within 24 hours.